
Kidney Beans Part 2: The Case of Renal Dysfunction
Editor’s Note: J Nile Barnes, PharmD, BCPS is an experienced clinical pharmacist and paramedic. He has over 30 years of experience in healthcare, including more than 20 years teaching paramedic and pharmacy school.
You may remember him from his prior extensive post series about heart failure (Part 1, Part 2, and Part 3), and if you don’t, be sure to add those posts to your reading list!
This particular blog set comes from years of teaching pharmacist interns about drug dosing and renal function testing. This topic is a standard topic on every APPE rotation he teaches. He has also presented this topic at a national clinical nursing convention.
Basically, we’re all lucky to be able to learn from his formidable brain!
The following is the second in a three-part series on renal function and drug dosing. The first part discussed the techniques for assessing renal function, this Part 2 will discuss the mechanisms for acute and chronic kidney dysfunction, and finally Part 3 will give some practical pointers for drug dosing with decreased renal clearance.
So let the learning continue!
We combined ALL of the posts in this Kidney series AND our Liver series into a single PDF. If you’d like a downloadable (and printer-friendly) version of this article, you can get one here
Remember Kidney Beans: Part 1? In that section, we discussed how renal function is measured and the issues associated with understanding the measurements. If this isn’t ringing any bells, please click the pretty blue letters above.
So, with part 1 behind us, let’s delve into what happens when all’s not sunshine and unicorns with the beans. I’m talking renal dysfunction, sometimes called uremia.
Generally, we have two big categories of renal dysfunction: acute kidney injury (AKI) and chronic kidney disease (CKD). Let’s begin by tackling AKI.
What Constitutes Acute Kidney Injury (AKI)?
The KDIGO (Kidney Disease, Improving Global Outcomes) group has guidelines that define AKI and give suggestions for evaluation and management. Although they were last updated in 2012, they are still quite applicable.
AKI is defined by KDIGO as any of the following:
Increase in SCr by at least 0.3 mg/dl (26.5 mol/l) within 48 hours;
Increase in SCr to at least 1.5 times baseline, which is known or presumed to have occurred within the prior 7 days;
Urine volume less than 0.5 ml/kg/h for 6 hours.
KDIGO also stages AKI based on severity.

The RIFLE classification is another way to define AKI (and renal dysfunction) promulgated by a different organization, the Acute Kidney Injury Network (AKIN). RIFLE is also a mnemonic:
Risk: SCr = 1.5-2 x baseline
Injury: SCr = 2-3 x baseline
Failure: SCr at least 3 x baseline
Loss of Function: at least 4 weeks
End Stage Renal Disease (ESRD): at least 3 months

KDIGO and AKIN at a renal conference. (Image)
Unfortunately, although both the KDIGO and RIFLE definition criteria have been validated, they don’t always necessary result in the same conclusion. For example, according to KDIGO, a SCr that is at least 1.5 times the baseline would mean Stage 1 AKI. However, according to the RIFLE criteria, that would only place the patient at risk for AKI. Confusing, right?
Well now that we’ve tried to define what AKI is (even if the powers that be are still leaving gray areas), let’s move into causes.
What Causes AKI?
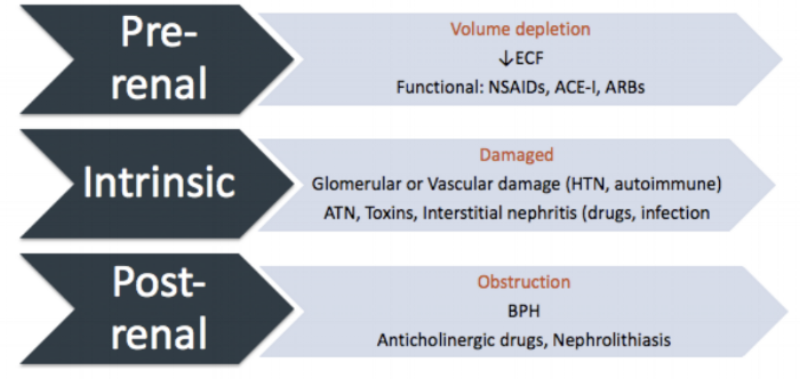
We can divide AKI into three subgroups: pre-renal, intrinsic, and post-renal disease.
Pre-renal AKI basically results from inadequate flow of blood to the kidney from shock or volume depletion. It seems obvious that if the blood does not get to the kidney, it cannot be adequately filtered. This can come from an actual decrease in blood volume (e.g., hemorrhage) or anything that makes it seem like reduced blood flow to the kidney.
For this latter scenario, drugs (e.g., NSAIDs, ACE-Is, ARBs) are the most common causes of this functional problem, but frankly a tumor pressing on the renal artery would have the same effect. Other causes include cardio- or hepatorenal syndromes and vasodilatory shock syndromes (e.g., septic shock).
To skip to the third subgroup, post-renal AKI results from an outlet obstruction causing urine flow to back up into the kidney. This back-up interrupts the normal gradients needed for filtration. The classic example of a bilateral physical obstruction causing post-renal failure is benign prostatic hyperplasia (BPH), but various tumor-causing cancers and neurogenic bladders are also implicated.

Hopefully this helps to make the geography of pre-, intrinsic, and post-renal issues clearer. (Image)
Nephrolithiasis (kidney stones) can cause unilateral obstruction, which can be significant in patients with underlying CKD or a single kidney.
Some drugs can decrease urine outflow as well. Classically, these are anticholinergic drugs (which, in some cases, we actually use therapeutically for patients with excess urine flow). Some other medications (e.g., acyclovir) can lead to development of intra-renal crystals if the patient is not adequately hydrated during therapy. These crystals function as obstacles just like classic kidney stones.
So then we can back up to the second (slightly beastly) subgroup. This is intrinsic AKI. This term just means there is something wrong with one of three things: the actual glomerular filtration system, the tubules (interstitium), or the vascular system within the kidney. Classically, intrinsic AKI is secondary to damage from excess pressure (usually from hypertension). But it can also occur from some drugs, as well as infection, toxins, autoimmune diseases, acute tubular necrosis (ATN), and others.
The two main types of intrinsic AKI that are most pharmacy-related are acute interstitial nephritis (AIN) and acute tubular necrosis (ATN). AIN is a bit like an allergic reaction in the kidneys, and as the name implies, involves inflammation. Beta lactam antibiotics are first-class culprits for this, but other medications like sulfonamides, NSAIDs, and PPIs have also been associated.
On the other hand, ATN is usually either related to ischemia or medications. When your hemorrhagic, hypotensive patient’s SCr does not improve even after adequate fluid resuscitation and stabilization of vitals, there’s a good chance the ischemic period may be causing ATN and consequent AKI. Medications toxic to the tubule cells, such as aminoglycosides, contrast agents, and amphotericin B, are likely sources of drug-induced ATN.

So for a thorough review of the causes of AKI… (Image)
For those of us who maybe don’t have eidetic brains and need simplification…
One mnemonic for remembering some of the important causes of AKI is VOID RIGHT:
A summary of the 3 main causes of AKI. See if you can match up the components of VOID RIGHT with these categories! (Also, ECF = extracellular fluid)
Vasculitis
Obstruction
Infection
Drugs
Renal artery stenosis
Interstitial nephropathy
Glomerular nephropathy
Hypovolemia
Thromboembolism
Now that we know more about the causes of AKI, how do we manage it when it happens?
Management of AKI
The best way to evaluate and treat AKI is to avoid it! Therefore, patients at high risk (i.e., those with multiple risk factors) should have risk factors mitigated when possible to prevent AKI. Periodically check SCr and urine output (UO) in patients at high risk.
When AKI does occur, promptly determine the severity and the cause when possible, with special attention paid to reversible causes (e.g., volume depletion). Realize it is likely that the cause is multifactorial. For example, consider the patient on an ACE-I for hypertension and ibuprofen for osteoarthritis who gets gastroenteritis, becomes anorexic, vomits, and becomes volume depleted.
Frequent SCr and UO measurements should be done when AKI is found. Most hospitalized patients should have daily measurements, but patients who deteriorate may need evaluation more often.
Several general strategies are recommended for managing AKI.

The shading indicates the importance of the intervention - the darker the shade, the more important KDIGO believes the intervention is. (Image)
Some of these are rather obvious; stopping nephrotoxins, ensuring adequate volume and perfusion pressure, and monitoring SCr and UO are straightforward interventions. Since hyperglycemia acts as an osmotic diuretic, this stresses the already injured kidney (oxidative stress) and can cause electrolyte disturbances. So glycemic control is important (as always!).
It is also fairly intuitive to avoid nephrotoxic interventions: avoid contrast media for radiological procedures. Many people believe MRI contrast is not nephrotoxic, but nephrogenic systemic fibrosis (NSF) is associated with gadolinium-based MRI contrast in patients with existing renal disease (i.e., you can’t get NSF without pre-existing renal disease). BTW, there are three types of gadolinium-based contrast agents, and the risks are not equal.
Other more specific recommendations from KDIGO include these:
Not using diuretics to prevent or treat AKI, except in the management of volume overload.
Not using low-dose dopamine to prevent or treat AKI.
Not using fenoldopam to prevent or treat AKI.
Not using atrial natriuretic peptide (ANP) to prevent or treat AKI.
Not using recombinant human (rh)IGF-1 to prevent or treat AKI.
Not using aminoglycosides for the treatment of infections unless no suitable, less nephrotoxic alternatives are available.
In patients with normal kidney function in steady state, aminoglycosides are administered as a single dose daily treatment regimens and monitoring if used for more than 48 hours.
Monitoring aminoglycoside drug levels when treatment with multiple daily dosing is used for more than 24 hours.
Using topical or local applications of aminoglycosides (e.g., respiratory aerosols, instilled antibiotic beads), rather than i.v. application, when feasible and suitable.
Using lipid formulations of amphotericin B rather than conventional formulations of amphotericin B.
In the treatment of systemic mycoses or parasitic infections, using azole antifungal agents and/or the echinocandins rather than conventional amphotericin B, if equal therapeutic efficacy can be assumed.
This first thing I notice from this list is to avoid nephrotoxic drugs.
Oh, and the second thing is to avoid nephrotoxic drugs.
The third thing is to avoid nephrotoxic drugs like aminoglycosides and conventional amphotericin B most (if not all) of the time.
Also, don’t do things that don’t have adequate evidence (small studies or animal models) like fenoldopam, ANP, or IGF-1.

Remember the evidence train? All aboard! (Image)
Oh, will a urine analysis (urinalysis) help determine the cause?

The 10 parameters on a basic urinalysis (UA). You can imagine how color blindness would basically make this impossible to interpret! (Image)
Likely, it will shed some light on the cause of the AKI. The most common way to test urine is for a urine dipstick test to be performed plus a microbiologic smear to be stained and viewed.
While there are several brands on the market, most have the ten tests seen in this colorful image.
These are colorimetric tests that are simply viewed by the clinician or laboratory personnel. It is extremely important that the person interpreting does not have significant color blindness.
I won’t create a tutorial here, but you can find one on Wikipedia.
The “micro” portion generally gives counts of white cells, red cells, bacteria, and urinary casts. It is extremely important to remember that a bacterial infection only exists if pyuria is present. Otherwise the presence of bacteria indicates colonization (aka asymptomatic bacteriuria). Other abnormalities may exist that give insights about the type of AKI.
So, what is a clinician to do when confronted with an AKI? Refer back to the KDIGO Consensus Guideline found above, remembering that common things are common and uncommon things are uncommon.
Look for common reversible causes including dehydration and infection and treat those before hunting for facinomas that do not have other clues. In other words, when you hear hoofbeats, looks for horses, not zebras!
Also remember to evaluate patients 3 months after AKI for resolution, new onset CKD, or worsening of pre-existing CKD.
If patients have CKD, manage these patients as detailed in the 2012 KDIGO Guideline (stay tuned for like another 2 seconds).
If patients do not have CKD, consider them to be at increased risk for CKD and care for them as detailed in Chapter 3 of the 2012 KDIGO Guidelines.
So now to explore this CKD business further…
What Constitutes Chronic Kidney Disease (CKD)?
Just like AKI, there are criteria for CKD. The most used come from KDIGO with their last guideline update in 2012 (still applicable). Their short definition is as follows:
CKD is defined as abnormalities of kidney structure or function, present for at least 3 months, with implications for health. CKD is classified based on cause, GFR category (G1–G5), and albuminuria category (A1–A3), abbreviated as CGA.

The CKD-EPI equation used for GFR estimation in KDIGO 2012 CKD guidelines. (Image)
Looking first at the GFR portion of the classification, it’s important to note that the general CKD-EPI equation for eGFR is used.
They also list several specific equations based on gender and SCr, as well as equations used for estimating eGFR using exogenous cystatin C and pediatric equations.
YOU CAN SPEND HOURS learning the subtleties of these various equations, and if you remember anything from Part 1 of this series, you will understand that these are all estimates of averages of varying renal function.
And while those who created them should be commended for the scientific inquiry and for creating what may be better estimations, ultimately, they are still estimations.
Now let’s factor in the second portion of the CKD definition, albuminuria.

The prognosis of CKD based on GFR and albuminuria. There’s a LOTTA red here. (Image)
So how does all that alarming red and increased risk play in? In addition to the guidelines using these risk levels to make recommendations for frequency of follow-up and monitoring, these levels also lend clues about the progression of the disease. Take a look at the conceptual model for CKD:

Risk is just the predecessor of many, many complications - up to and including death. (Image)
You should see that some things are reversible (left pointing arrows), but most are not easily reversible. This means that many patients with CKD will continue to progress toward kidney failure (right pointing arrows), which has implications for drug therapy (stay tuned for Part 3 of this series). There are also potential complications at every level in the conceptual model (diagonal lines), which require management and attention.
Yikes! Well, now that we’ve taken a look at the complicated version of defining and staging CKD, let’s simplify.
The American College of Physicians (ACP) released its own 2013 guidelines for CKD management. In this version, there is a CKD staging system based on just GFR rather than also factoring in albuminuria. Please note ACP’s CKD stages 1 and 2 require concomitant kidney damage. In other words, having an eGFR of at least 90 does not mean you have stage 1 CKD unless you also have demonstrable kidney damage.

Simplified GFR-only staging of CKD according to the American College of Physicians. (Image)
Please notice that even with a GFR of at least 90 mL/min/1.73m2, you can still have CKD! CKD is not simply decreased GFR, and decreased GFR is not simply CKD.
The other really hard part here is that it is an insidious silent disease initially. Stages 1-3 usually do not have symptoms, and late stages may have only subtle clues.
On that note…let’s have a side bar.
I want to emphasize that, for an initial evaluation, it can be difficult to figure out if the patient has an acute injury alone, chronic disease alone, or acute-on-chronic renal disease. If the renal dysfunction does not resolve in three months, then a chronic disease component exists.
Example: Mr. Jones is a 72 YOM who presents with an eGFR ~ 35 mL/min/1.73m2. He has started on ibuprofen 400 mg tid for osteoarthritis and has been on lisinopril 30 mg daily for the past 20 years. (He buys the lisinopril over the internet to save money and does not see his physician regularly). He recently had a “nasty cold” and was not eating or drinking for “several days.” He appears clinically dehydrated and “tilts” when orthostatic vital signs are done.
Does he have:
a) No kidney issues
b) AKI alone
c) CKD alone
d) AKI on top of pre-existing CKD
e) b, c, or d could be correct
The only answer that is clearly wrong is A! But you really can’t tell if he is just at his baseline with pre-existing CKD and the three potential hits to his kidneys haven’t done anything. Versus this a significant AKI and after treatment his renal function will return to normal. Versus this a situation of CKD with a low eGFR that has been pushed lower by the AKI.
Following this man’s renal function for the next three months will reveal the answer. If he resolves completely within days of therapy, this was AKI alone. If he partially resolves, this is AKI on top of CKD (historically called Acute on Chronic Renal Failure). And if there is no resolution in three months, this is CKD. Please note that the CKD could be new or pre-existing.
<End Side Bar>
Management of Chronic Kidney Disease
It is important to realize that the early stages of CKD are not benign even if they are often asymptomatic. Remember how we said in the conceptual model of CKD above there were complications associated with every level of the disease?
Even the early stages are associated with increased mortality, cardiovascular disease, fractures, bone loss, infections, cognitive impairment, and frailty. And while there is no specific pharmacotherapy for CKD, it is important that these other conditions are addressed. In their same 2013 guidelines, the ACP recommends ACE-Is or ARBs for hypertension in these patients and statins for hyperlipidemia.
Pharmacists can help manage CKD by optimizing drug therapy that slows progression of the disease and also prevents AKI. In addition, the pharmacist must account for this disease and its effects on pharmacokinetics when dosing drugs. Patients with low eGFR, on dialysis, and transplant recipients need special attention with regards to nephrotoxic drugs.
For CKD stages 4 and 5, the added problems of preparing for Renal Replacement Therapy (RRT) or performing RRT are added. RRT is either chronic dialysis or renal transplant. Dialysis can be either hemodialysis (HD) via a dialysis machine or peritoneal dialysis (PD).
Because causes of CKD are a heterogenous mix, there is no single set of interventions that every patient should get. However, CKD is an almost “inexorably progressive” disease with important cardiovascular (CV) implications, so evaluating and managing CV risk is always required. CV death in CKD is more common than progression to dialysis (25 times more likely than progression to kidney failure).
There are, however, some general recommendations to consider:
Use of RAAS inhibitors should be considered.
Avoid postural hypotension with vasoactive drugs.
Using both an ACE-I and ARB is NOT recommended.
Careful monitoring to see that patients are at BP goals. (Goals vary by organization but </= 130/80 is a reasonable goal.)
Periodic urine albumin checks.
Avoid AKI.
With low eGFR (<30), decreasing protein intake to less than 0.8 g/kg/day.
Avoiding high protein intake (>1.3 g/kg/day) in adults with risk of CKD progression.
Targeting an HbA1C ~7% to prevent progression of diabetic kidney disease for most patients (may be more liberal in patients at high risk of hypoglycemia or with significant co-morbidities).
Decreasing salt intake to less than 5 grams of NaCl daily (<2 grams of sodium).
Physical exercise as tolerated for CV health - preferably 30 minutes daily, five times a week.
Smoking cessation.
Dietary advice from an expert dietician regarding sodium, potassium, phosphates, and protein intake.
Monitor for and diagnose anemia.
Measure calcium, phosphate, PTH, and alkaline phosphatase in patients with eGFR <45 to inform a baseline and respond as needed.
If serum bicarbonate levels are less than 22 mmol/L consistently due to CKD, oral bicarbonate supplementation should be considered.
All healthcare providers should know how to deal with CKD and its complications. Not all patients with CKD should be referred to a nephrologist just as not all diabetic patients are referred to endocrinologists.
So, like in part 1, let’s look a couple of cases.
Case 1
BillyRayJoeBob (I am a southern redneck, so I can pick on this group) is dragged into the clinic by his wife for new onset confusion and shortness of breath. He was last seen in the clinic 4 years ago for a hand injury, and they managed to get some blood work done at the time since he rarely follows up and often refuses tests (“I am fine.”)
His baseline SCr was 0.9 mg/dL, and BUN was 12 at the time. He is now 44 years old, 6 feet tall, and weighs about 180 pounds (weight was 170 at the last visit). At previous visit, the CrCl was estimated at 119 ml/min and the eGFR was >60 ml/min/1.73 m2 . On this visit his SCr is 5 mg/dL, and his BUN is 219.
His is mildly confused. His wife tells you he has been drinking heavily lately, not eating well for months, and constantly complains of knee, hip, and back pain. He agrees and tells you he has been taking both ibuprofen and Motrin for the pain. His physical exam is remarkable for fine crystals being left behind where he sweats. He has bilateral basilar rales and 3+ edema up to his knees bilaterally. Today’s CrCl is about 19, and eGFR is likely about 12. Oh, and his potassium is 9.0 mg/dL. What is going on?
He obviously has severe kidney dysfunction. However, do we know enough to decide if it is AKI or CKD? Is there any emergent or urgent therapy he needs?
Let’s tackle the second question first. How do we know when to initiate dialysis?
The criteria for emergent dialysis are: AEIOU.
A - intractable acidosis
E - electrolyte, severe disturbance (K+, Na+, Ca++)
I - intoxicants (methanol, ethylene glycol, Li, ASA)
O - intractable fluid overload
U - uremic symptoms (nausea, seizure, pericarditis, bleeding)
Dialyze early. Keep BUN <100 mg/dL and SCr <10 mg/dL.
So, for our guy, yes, there is an emergent need for hemodialysis!
1) His potassium is dangerously high,
2) He has fluid overload, and
3) His BUN is so high he is leaving a uremic frost on his skin.
So, he is emergently dialyzed that day and every day for the next 3 days. His laboratory data returns to near normal (SCr =1.6, BUN 20), and his peripheral and pulmonary edema resolve. He no longer needs dialysis. His NSAIDs (remembering he was double dosing them) are stopped, and he is diagnosed with a rheumatological condition explaining his joint pain. His pain is now controlled with narcotics and steroids, and he will have appropriate follow up for his pain.
So, this is clearly a pre-renal AKI. Likely caused by afferent renal arteriolar vasoconstriction that lowers glomerular filtration pressure. NSAIDs, by inhibition of prostaglandins and bradykinin, produce vasoconstriction of the afferent renal arteriole and reduce the ability of the kidney to regulate (increase) glomerular blood flow.
What isn’t so clear is the question of CKD. Has he done permanent damage to his kidneys?
Time will give us the answer.
If his SCr and eGFR stabilize at normal levels within 3 months, then, “No, he does not have CKD.” Just a very acute AKI. If his SCr and eGFR do not return to normal (I would also look at albuminuria), then he has CKD.
Case 2
BillyJeanBobbySue presents with sepsis to the ED. She is hypotensive (BP 78/48, HR 118 ) on admission and is fluid resuscitated in the ED with 3 liters of LR. She is simultaneously treated with broad spectrum antibiotics (vancomycin and piperacillin/tazobactam, aka VPT) until her blood culture returns.
Her SCr in the ED on arrival was 1.6, and a BUN was 20. Twelve hours later her BP is 110/64, and HR is 84 at rest. Her SCr is 1.9, and BUN is 24. Twelve hours after that her SCr is 2.4 and BUN is 30, although her vital signs are still stable. What is going on?
This is likely a pre-renal AKI due to decreased renal perfusion from the septic shock. But it could easily be complicated by intrinsic injury from both hypotension and medications. The mechanism of injury from VPT is not clear, but interstitial nephritis and proximal tubule injury have both been implicated.
Is there CKD? I would bet not. Her initial BUN/SCr were not greatly elevated, reflecting the previous 24 hours of renal function. When she went into septic shock, her kidneys were not perfused well for a time. Then when they were perfused, she received VPT.
Vancomycin has long been known to cause AKI. Decades ago it was due to impurities in the preparation. In fact, the nickname for vancomycin was ”Mississippi Mud” owing to the brown impurities in the IV bag. Since 2011, there have been many case reports and studies of VPT therapy having a greater incidence of AKI than vancomycin alone. For example, see here, here, and here, just to name a few! In response, some authors have suggested that the broad spectrum “work horse” be changed from VPT to vancomycin plus cefepime (VC).
It is entirely probable that this is only an AKI and that it will resolve with continued good perfusion of the kidney and minimization of nephrotoxic drugs.
In BillyJeanBobbySue’s case, her blood cultures come back with E. coli, a Gram-negative rod, so vancomycin can be discontinued.
Alright. Alright. Alright.
In Kidney Beans Part 1, we looked at ways to measure renal function and the perils and pitfalls of making assumptions that are not true. Here in Kidney Beans Part 2, we have discussed AKI and CKD. In Kidney Beans Part 3, we will look at the implications for drug dosing in these clinical scenarios.
We combined ALL of the posts in this Kidney series AND our Liver series into a single PDF. If you’d like a downloadable (and printer-friendly) version of this article, you can get one here