
When Good Drugs Go Bad: Beta-Blocker Toxicity
Joe’s Note: Welcome back to our series of “When Good Drugs Go Bad.” We first kicked this series off with the world's most common drug overdose: acetaminophen. If you haven’t done so, go ahead and give that a read. You’ll thank me the next time you’re asked about N-acetylcysteine.

For today’s topic, I figured it’d be good to talk about beta-blocker toxicity considering how commonly used they are. Heart failure, atrial fibrillation/flutter, hypertension, akathisia, migraine prevention, performance anxiety, thyroid storm, essential tremor, and portal vein hypertension…just to name a few indications. And let’s be real, it’s all fun and games until you’re asked to verify an insulin infusion running at 100 units/hour for a beta blocker overdose. (Image)
So, to avoid this scare, let’s get right into it.
How Do Beta Blockers Work?
To understand the toxicity of a drug, it’s important we first review how a drug actually works. Once we understand the mechanism of action, the toxicity portion will make more sense. Now, we’re not going to focus on the nitty gritty details of beta blockers in this post, but luckily for you, we have a previous post that goes over everything you need to know about beta blockers. This includes how they work, when they’re used, different clinical pearls, indications, and much more. For now, I’ll give you a quick refresher.
Remember our autonomic nervous system? And how our autonomic nervous system is further broken down into the sympathetic (“fight or flight”) and the parasympathetic (“rest and digest”) nervous systems? Oh, and remember those alpha and beta receptors? No?
Okay fine. Let’s review.
Alpha-1 (A1)
Location: blood vessels
Agonist Effect: vasoconstriction
Beta-1 (B1)
Location: heart
Agonist Effect: increase in heart rate (chronotropy) and heart contractility force (inotropy)
Beta-2 (B2)
Location: Primarily lungs
Agonist Effect: bronchodilation
So now moving on to beta blockers. As the name suggests, they primarily block beta receptors (duh). Some are cardioselective while others are non-selective. The cardioselective ones only block B1 receptors (found in the heart), while the non-selective ones can block B1, B2, and even A1. And in case you need a quick refresher on which ones block what:

And yes, I even color-coded them for you. The ones in blue have alpha-adrenergic blocking properties as well as antagonizing both B1 and B2. The ones in orange have intrinsic sympathomimetic activity.
Signs & Symptoms of Beta Blocker Toxicity
The clinical features of beta blocker toxicity are pretty straightforward now that we understand how beta blockers work. If we antagonize B1, we get reduced heart rate and decreased cardiac output. If we antagonize B2, we get bronchoconstriction. If we antagonize A1, we get vasodilation. So, how do overdose patients present?
Cardiovascular (most common): bradycardia, hypotension, heart block, cardiogenic shock
Central nervous system (if severe toxicity): altered mental status, seizures, coma
Respiratory: bronchospasm, respiratory depression
Metabolic & Endocrine: hypoglycemia (due to inhibited glycogenolysis and gluconeogenesis), metabolic acidosis, hyperkalemia
Other: cold, clammy skin, hypothermia
Treatment Algorithm for Beta Blocker Toxicity
A lot of meds can be used to treat beta blocker toxicity. Sometimes it can feel overwhelming with how many options we have. I promise we’ll review each treatment one by one. But before I throw all these meds at you, I figured it might be nice to give you a visual of the treatment algorithm first. Hopefully this will help simplify the treatment:

What’s the Role of Activated Charcoal in Beta Blocker Toxicity?
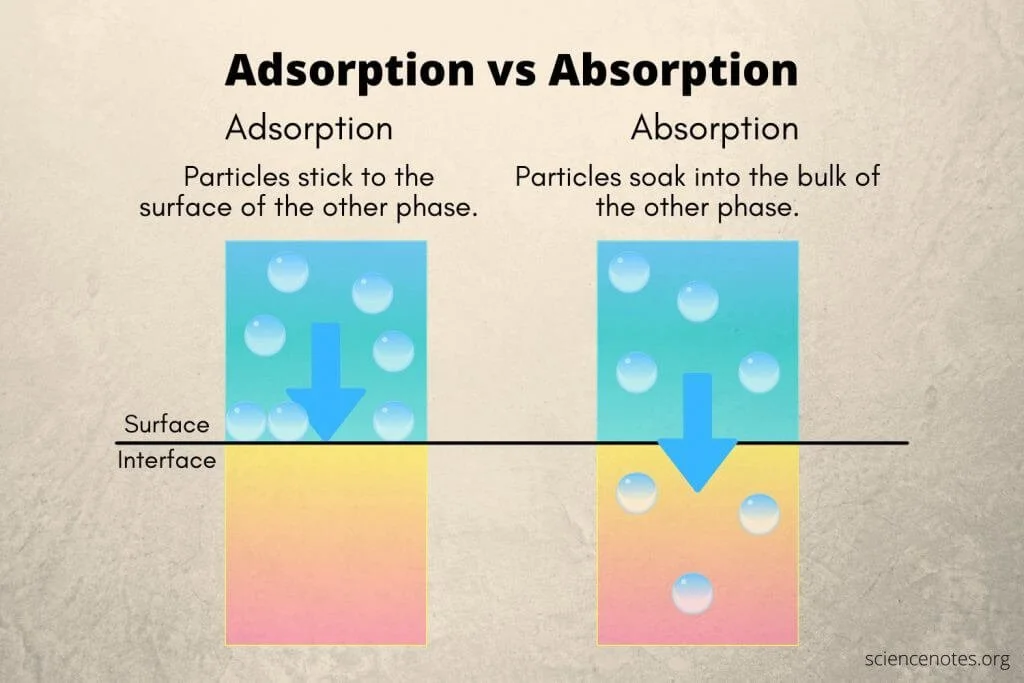
Activated charcoal is hit or miss. Different studies tell you different things. It’s not a specific antidote for just beta blockers. It can really be used for the intoxication of any oral medication. How?
It adsorbs toxic substances and inhibits GI absorption to prevent or limit systemic toxicity. (Image) The sooner you give it the better the outcome. The longer you wait, the more a drug gets absorbed, and the less effective the activated charcoal is.
In fact, there’s this 2005 study that tested the mean reduction in drug absorption following a single dose of ≥50 g activated charcoal. Here are the results:
If given within 30 minutes after ingestion: 47.3% reduction
If given at 60 minutes after ingestion: 40.07% reduction
If given at 120 minutes after ingestion: 16.5% reduction
So, if your patient presents within ~2 hours of a beta blocker ingestion, you can potentially administer 25 to 100 g of activated charcoal. Also don’t forget that you have a longer administration window with extended release (ER) formulations (like metoprolol succinate). In that scenario, you may still administer activated charcoal even 4-6 hours after ingestion.
Atropine for Beta Blocker Toxicity
Atropine is one of those drugs that’s usually not very effective…but also doesn't have much risk. Kinda one of those “eh, let’s give it a try” drugs. I mean if you go to LexiComp, it literally says, “Note: atropine may not be effective for type II second-degree or third-degree heart blocks.”
I am not making this up. You can go and check for yourself. But either way, it’s still going to be a drug that we give for beta blocker toxicity.
If you don’t remember, atropine is an anticholinergic agent. It blocks the action of acetylcholine at the parasympathetic (rest & digest) nervous system. So what effects do you get? The opposite of rest and digest. I.e., increased heart rate, cardiac output, dryness, constipation, decreased urination, etc. The main effect we’re aiming for in the scenario of beta blocker toxicity is the increase in heart rate. Everything else is an unwanted side effect (but can be dealt with later).
Atropine can be given IV, IM, or IO. IV is always going to be preferred as long we have intravenous access. Regarding dosing, give 1 mg of IV atropine every 3 to 5 minutes as needed until symptoms resolve or heart rate is stabilized. Don’t forget, there is a max of 3 doses (max total dose: 3 mg). After that, we move on to other agents.
Glucagon for Beta Blocker Toxicity
This one always confused me. Glucagon is a hormone produced by the pancreas that plays a crucial role in regulating blood sugar levels. When serum glucose drops too low, glucagon is released, which then stimulates the liver to release stored glucose into the bloodstream. It pretty much is the perfect counter for insulin.
But its use in this setting is slightly confusing. How does increasing serum glucose have anything to do with beta blocker toxicity?

Yes, glucagon increases glucose release from the liver. But there are also specific glucagon receptors on the heart (surprise, surprise!). (Image) When glucagon binds to these receptors, it stimulates the production and release of cAMP. Increased cAMP levels lead to increased calcium influx into myocardial cells, which in turn enhances myocardial contractility, heart rate (positive chronotropy), and atrioventricular (AV) conduction. This directly reverses the effects of beta blocker toxicity.
Pretty neat, right?
Glucagon should only be given intravenously when used to treat a beta blocker overdose. Dosing generally ranges from 2 to 10 mg as a bolus with a potential to repeat after 10 to 15 minutes. If clinical response is achieved with the bolus, start a continuous infusion at 2 to 5 mg/hr, and titrate the rate to achieve adequate hemodynamic response.
Calcium for Beta Blocker Toxicity
Calcium administration is usually more integral for a calcium channel blocker overdose; however, it may help with beta blocker toxicity as well. How? Great question.
Like we talked about earlier, beta blockers inhibit B1 receptors in the heart → lead to decreased cAMP → reduced intracellular calcium influx → leads to bradycardia, hypotension, negative inotropy, and cardiogenic shock.
So if we give intravenous calcium, this should hypothetically increase intracellular calcium influx, which would counteract the negative cardiac effects caused by beta blockers (particularly the negative inotropy). Calcium does not directly reverse receptor blockade. Instead, it overrides the downstream effects by flooding the myocardium with calcium, helping restore vascular tone and cardiac output.
In terms of calcium products, you can either use calcium chloride or calcium gluconate. Remember, calcium chloride is approximately three times more concentrated in elemental calcium than calcium gluconate (1 g of calcium chloride = 270 mg elemental calcium; 1 g of calcium gluconate = 90 mg of elemental calcium).
Therefore, calcium chloride generally needs to be pushed slower and through a central line if able. How do you choose a calcium product? Simple. Give calcium chloride for severe beta blocker toxicity with major hemodynamic instability and calcium gluconate for more mild/moderate beta blocker toxicities. In terms of dosing:
Calcium chloride: 1 to 2 g over 5-10 minutes; may repeat every 10-20 minutes for 3 to 4 additional doses or initiate a continuous infusion of 20 to 40 mg/kg/hour titrated to improve hemodynamic response
Calcium gluconate: 3 to 6 g over 5-10 minutes; may repeat every 10-20 minutes for 3 to 4 additional doses or initiate a continuous infusion of 50-120 mg/kg/hour titrated to improve hemodynamic response
Vasopressors for Beta Blocker Toxicity
If you’ve given IV fluids, atropine, glucagon, and calcium but your patient remains hemodynamically unstable, then it’s time to add a vasopressor. This one is pretty self explanatory in my opinion. Beta blockers inhibit B1, B2, and maybe even A1 receptors (depending on the agent). Vasopressors agonize those same receptors and therefore counteract the toxic effects of beta blockers.
Now we’re not going to go super in-depth on how vasopressors work and all that. We have an entire post on vasopressors. Choosing a vasopressor to reverse beta blocker toxicity is generally institution specific as there is no clear guidance on which pressor to utilize first. That being said, we should still use our clinical judgment when determining which vasopressor to start for our patients.
Like we’ve talked about, not every beta blocker works the same. For example, metoprolol is cardioselective and only inhibits B1 receptors. However, carvedilol is not as selective and usually inhibits B1, B2, and
A1 receptors. When picking a vasopressor, try to use an agent with a mechanism of action that would counteract the effects of whatever beta blocker toxicity we’re treating.
For example, epinephrine agonizes B1, B2, and A1. So if we have a carvedilol overdose, then epinephrine might be our best agent. On the other hand, phenylephrine only agonizes A1 and has no effect on beta receptors. So in this situation, phenylephrine would NOT be the best choice for treating beta blocker toxicity.
High-Dose Insulin Euglycemia Therapy (HIET) for Beta Blocker Toxicity
If you’ve given all the above agents and nothing seems to be helping then you move on this super scary high-dose insulin therapy. High-dose insulin euglycemia therapy (HIET) is very confusing and startling at the same time. I mean, you usually start this infusion at 1 unit/kg/hour but can go up to >10 units/kg/hour.

Let’s just take a moment to talk about how crazy that is. That means if you have a 100 kg patient, there is a possibility that you could crank that infusion to >1000 units/hour. That’s legit madness, folks. (Image)
Before we go into the crazy dosing, let’s talk about how high dose insulin helps with beta blocker toxicity. I’m going to preface this by acknowledging that the mechanism is not completely understood. But there are multiple theories on how high-dose insulin with dextrose may help with beta blocker toxicity. These include:
Shifts myocardial fuel preference: in a stressed state, the heart’s preferred energy source shifts from free fatty acids to glucose. Beta-blocker toxicity impairs the heart’s ability to use available glucose and fatty acids. So....
Give high dose insulin with dextrose → increase the uptake and utilization of glucose by the heart cells → allows the heart to function more effectively
Positive inotropic effects:
Stimulates the phosphatidylinositol-3-kinase (PI3-K) pathway → leads to increased intracellular calcium availability
Increases the activity of calcium-dependent ATPase in the sarcoplasmic reticulum → improves calcium inflow into mitochondria → allows for stronger contractions
Long story short, HIET with dextrose provides the heart with more glucose and calcium leading to more “food” and “power” for the heart to pump more effectively. This in turn leads to positive inotropy and reversal of beta blocker toxicity.
Before we go into the dosing of HIET, let’s review some really important points since this is a very high risk medication. For starters, you need to get baseline labs before starting HIET. Baseline labs should include:
Venous blood gas (VBG)
Blood glucose
Serum potassium
Serum magnesium
Serum phosphorus
Speaking of labs, remember that IV insulin pushes potassium back into the cells and can cause severe hypokalemia if not treated appropriately. This is very dangerous and can be life-threatening if not accounted for. So remember to always correct hypokalemia prior to initiation of insulin therapy. And in terms of glucose, a dextrose infusion will always be co-administered with HIET. Your goal is to keep the blood glucose between 150-250 mg/dL.
Okay now onto dosing. Each institution has slightly differing protocols regarding HIET. So make sure you review your hospital’s protocol and get familiar with it. But to avoid any bias, I will use the recommended dosing found on LexiComp.
IV (U-100): 1 unit/kg bolus, followed by a continuous infusion at 0.5 to 1 unit/kg/hour titrated to clinical response.
Higher doses (e.g., boluses of up to 10 units/kg and continuous infusions of >10 units/kg/hour) have been administered with good outcomes and minimal adverse effects.
Once hemodynamic parameters have stabilized, gradually decrease insulin infusion.
Generally speaking, the benefits of HIET are seen pretty quickly with most cases improving within 30-60 minutes of initiation. So if the insulin infusion has been running for a long time with no benefit, it might be time to move onto our next agent.
I told you there were a lot of treatment options, didn’t I?
Lipid Emulsion Therapy for Beta Blocker Toxicity
On to our last-line agent. This is when everything isn’t working and your patient has failed IV fluids, activated charcoal. atropine, glucagon, calcium, multiple vasopressors, and HIET. Very few patients ever reach this point. But if they do, you have one last option: lipid emulsion therapy.
However, there is a caveat here. Intravenous lipid emulsion (ILE) can’t be used to treat all beta blocker overdoses. ILE is only beneficial in treating lipophilic beta blocker toxicities such as propranolol, metoprolol, and labetalol. It is unlikely to be effective when treating non-lipophilic beta blockers such as atenolol. Why is that?
Because when you give intravenous lipid emulsion, it creates an expanded, lipid-rich phase within the bloodstream. Highly lipophilic drugs preferentially partition or are “sequestered” into these circulating lipid droplets. This decreases the concentration of the free drug available to bind to target tissues. In addition, the emulsion then acts as a “lipid shuttle” and transports the bound drugs from the heart to sites of metabolism and excretion such as the liver.
So, if the beta blocker of concern is hydrophilic, then none of this would occur, and there would be no benefit. Bah.
In terms of dosing, most protocols recommend giving an IV bolus of 1.5 mL/kg of a 20% lipid emulsion solution over one minute. If there is no response, the same dose can be repeated every 3 to 5 minutes for a total of three bolus doses.
The tl;dr of Beta Blocker Overdose
We covered quite a lot of information today. Kudos to you if you made it all the way to the end. In case you didn’t, here’s a quick tl;dr version of everything we talked about…
Beta-blockers primarily block B1 and B2 receptors. Certain beta blockers (e.g., carvedilol, labetalol) antagonize A1 receptors in addition to B1 and B2.
Patients with beta blocker toxicity commonly present with bradycardia, hypotension, cardiogenic shock, altered mental status, bronchospasm, hypoglycemia, and hypothermia.
A step-wise approach to treating beta-blocker toxicity would be IV crystalloid fluid (if hypotensive) → activated charcoal (if appropriate) → atropine → glucagon → calcium → vasopressors → high-dose insulin euglycemic therapy → lipid emulsion.
Activated charcoal may be administered to patients that present within ~2 hours of beta blocker ingestion. Atropine is an anticholinergic agent that is known to increase heart rate by blocking the parasympathetic nervous system (“rest & digest”). A maximum of 3 total doses of atropine may be administered when treating beta blocker toxicity.
IV glucagon treats beta blocker toxicity by stimulating the production and release of cAMP leading to increased calcium influx into myocardial cells. Increase in intracellular calcium enhances myocardial contractility, heart rate, and AV conduction.
Either calcium chloride or calcium gluconate may be administered to treat beta blocker toxicity. Calcium chloride has 3x more elemental calcium than calcium gluconate and generally requires a central line. Calcium gluconate can be administered peripherally but generally requires higher dosing than calcium chloride.
There is no one preferred vasopressor for treating beta blocker toxicity. However, it is always recommended to administer a vasopressor with good B1, B2, and A1 activity, such as epinephrine.
High-dose insulin euglycemia therapy (HIET) may be beneficial in treating beta blocker toxicity as it provides the heart with more glucose and calcium, which in turn leads to positive inotropy and chronotropy. Before starting HIET, it is recommended that baseline labs be collected, including VBG, blood glucose, serum potassium, magnesium, and phosphorus. HIET must always be administered with a dextrose infusion to ensure blood glucose remains between 150-250 mg/dL.
Lipid emulsion therapy may be used as a last-line agent for treating beta blocker toxicity, but it is only beneficial when treating lipophilic beta blocker overdoses, such as propranolol, metoprolol, and labetalol. It is unlikely to be effective when treating non-lipophilic beta blockers, such as atenolol.