
The tl;dr Pharmacy Journal Club: Voxelotor in Sickle Cell Disease (HOPE Trial)
Courtney’s Note: If you’ve been paying attention to the news this week, you probably saw that Pfizer is voluntarily withdrawing Oxbryta (voxelotor) from the market due to safety concerns. Oxbryta was approved by the FDA under the accelerated approval pathway in 2019 for the treatment of sickle cell disease in adults and pediatric patients 12 years of age and older, and for those 4 to 11 years of age in 2021. Unfortunately, the manufacturer reported a higher rate of vaso-occulsive crises and more deaths in the Oxbryta treatment group compared to the placebo group in postmarketing studies. Because of this, the manufacturer has determined that the benefits of treatment do not outweigh the risks.
Even though this treatment is no longer available, we wanted to leave this article on our site because 1) It is a well-thought out journal club that may help guide you in writing your own, and 2) It highlights the risks of the accelerated approval pathway. The program is intended to allow for earlier approval of drugs that treat serious conditions and fill an unmet medical need based on a surrogate endpoint. Unfortunately, many of these drugs do not show benefit (or, in this case, may actually be deemed harmful) in postmarketing studies and are removed from the market. Hmmm… Sounds like a good topic for a future article, no?
Steph’s Note: This week, we’re going to change gears from all the recent talk about diabetes to another commonly-encountered general medicine topic: sickle cell anemia. And we’re going to do it journal club style (no, not Gangnam style, but good try).
I always tell my students on rotation that I LOVE journal club. Not because I like to pimp learners about some obscure statistical test (sheesh, I’m not that mean!), but because I always walk away from the discussion having learned something new. Whether it’s a perspective that I hadn’t considered during my first once over, or it’s a treatment that I’d never heard of, I love to hear other people’s thoughts about articles. So, putting the soapbox away, all I’ll say on this topic is please PLEASE read articles and chat together. Chat with your classmates, your preceptors, your boss at work - you will gain more if you talk!
One person I chatted with about this particular journal club is Cassandra Schmitt. She’s a PY4 pharmacy student at Virginia Commonwealth University in Richmond, Virginia, and in addition to our mutual love of horses, she is interested in pursuing a residency in critical care or emergency medicine. Right now, though, she’s currently learning about (and rocking, I might add) the extreme variety of internal medicine, which is where this sickle cell journal club topic arose. Cassie was chill enough to let me pull a two-fer here: read and analyze the article for discussion both with her AND with you. Plus, Cassie made some fantastic points during our chat, so I had to give her some credit here :)
Our article for this discussion is the New England Journal of Medicine’s June 2019 publication, “A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease.”
But before we can dive into journal club, we gotta at least do a quick review of sickle cell anemia.
What is Sickle Cell Anemia?
Sickle cell anemia is actually just one subset of sickle cell disease, which is an umbrella term that includes multiple variants of an inherited blood disorder. Make sure to also check out the tl;dr overview of sickle cell disease here.
The American Society of Hematology estimates that up to 100,000 people in the United States have sickle cell disease, with Blacks and Hispanics being the most highly affected. The CDC currently reports 1 of 365 Black and 1 of 16,300 Hispanic newborns are affected by sickle cell disease.
Although the median survival time has improved to be over 50 years in the US, it’s still estimated that people with this disease have a shortened life expectancy by about 30 years compared to non-sickle cell counterparts. And that’s regardless of access to medical care. And it’s certainly not reflective of sickle cell disease in other areas of the world, where the life expectancy is much shorter.
Which is CRAZY. Consider the difference between living to 50 and living to 75 - 80.
So what on earth is going on in these patients with sickle cell disease that is causing this shortened life expectancy?
What all of these versions have in common is the presence of hemoglobin S.
Ok, let’s take a step back.
What is Hemoglobin?
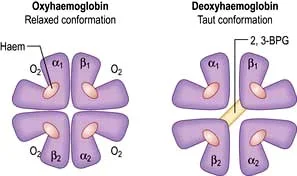
Basically, I think of it like this: when oxygen’s bound to hemoglobin, all the subunits get all relaxed and friendly, and the gaps between the subunits narrow. When there’s no oxygen, the subunits are farther apart because there’s the added need for stabilizing bonds. (Image)
Normal adult hemoglobin (HbA) is the protein found in red blood cells that is responsible for carrying oxygen.
Each hemoglobin molecule has 4 subunits (the “quaternary structure”). Two of these are alpha polypeptide chains, and the other 2 are beta polypeptide chains.
Each of the 4 subunits (2 x alpha, 2 x beta) has an iron-containing heme component that binds oxygen. Therefore, by simple math, each hemoglobin molecule can bind 4 molecules of oxygen for transport.
Oxygenated hemoglobin has a different configuration than deoxygenated hemoglobin. When hemoglobin binds oxygen, the entire molecule contracts; on the other hand, deoxygenated hemoglobin has additional stabilizing bonds between the 2 beta subunits, which make it more rigid and taut.
What is Hemoglobin S?
Hemoglobin S (HbS) is an inherited variant of HbA. A single gene mutation affecting the beta hemoglobin subunit is responsible for this version of hemoglobin, and people with sickle cell disease can be homozygous for the HbS gene (true sickle cell anemia phenotype = the most severe type) or they may be heterozygous if they only inherit one.
Of course, people who are heterozygous may not inherit normal HbA with their HbS gene. Rather, they may inherit some other abnormal hemoglobin gene, including hemoglobin C or a type of beta-thalassemia. Any of these other abnormal hemoglobin genes in combination with HbS may produce alternative phenotypes of the disease.
It’s also possible to inherit the HbS gene with a normal HbA gene, which is referred to as having sickle cell trait. People with sickle cell trait usually have a normal phenotype without clinical manifestations of any sickle cell disease.
So what’s the big deal about having this abnormal HbS?

(Image)
Hemoglobin S is unique because, when it loses its attached oxygen molecules, it polymerizes. Aka multiple HbS molecules bind to each other, making long, insoluble strands.
These long strands of bound hemoglobin distort the shape of the red blood cells to be long and thin. Hence, the cells are “sickle” cells, named for the farm tool of a similar shape. These abnormal erythrocytes are also less flexible than their round, plump, normal counterparts.
Complications of Sickle Cell Anemia
Because of their abnormal shape, decreased flexibility, and changes in the adhesion molecules on the outside of the erythrocyte, sickle cells with polymerized HbS can cause a hemolytic anemia and clog up blood vessels.
This can lead to vaso-occlusion, which in turn can precipitate tissue ischemia, fatigue, and organ damage.
Sickle cell pain crises are common manifestations of the tissue ischemia and damage that comes from vaso-occlusion, but complications of sickle cell anemia can be more widespread than pain alone. Think of all the organs and processes that require blood flow…
Complications of sickle cell disease can include the following systems (with examples):
Cardiopulmonary (acute chest syndrome, pulmonary hypertension, dysrhythmias)
Nervous (strokes, retinopathy, pain)
Musculoskeletal (avascular necrosis)
Urogenital (priapism, renal failure)
Reticuloendothelial (hyposplenism), and
Gastrointestinal (mesenteric vaso-occlusion, hepatopathy).
Treatment of Sickle Cell Disease
When this article was originally written in 2019, there were really only two FDA-approved medications for the treatment of sickle cell disease: hydroxyurea and L-glutamine.
Luckily, more treatment options (crizanlizumab, Casgevy, and Lyfgenia) have emerged in recent years, but at the time very few were available.
Hydroxyurea inhibits the cell cycle by inhibiting the process of DNA synthesis. At the dosing used for sickle cell disease, this intermittent inhibition is thought to reflexively result in recruitment of erythrocyte progenitors, which contain higher levels of the more protective form of fetal hemoglobin, HbF.
What, you say?! Yet another form of hemoglobin? Yup.
Mini teaching moment: fetal hemoglobin is exactly that - it’s the type of hemoglobin that is found in developing babies. HbF (which is found in F cells) is thought to be protective by lowering levels of HbS and slowing the HbS polymerization process. So HbF is helpful in sickle cell patients, which is why hydroxyurea is used (even though it seems so counterintuitive to use a cytotoxic drug in an already anemic person!).
Some patients don’t tolerate hydroxyurea very well. It can cause bone marrow suppression (we do use it to treat leukemia, after all), which may require holding and/or reducing doses. For some of our patients, the associated alopecia and eczema are deal breakers, and it also shouldn’t be used during pregnancy. For many patients of child-bearing age, this adds another factor for consideration.
L-glutamine (Endari) is an amino acid powder that requires mixing into a smoothie…of sorts. Each 1-3 packet dose is mixed in 240mL of a beverage, and this is consumed twice daily.
Even though that doesn’t sound too bad at first, how many people want 2 medication smoothies a day? Every day? Not to mention it is very expensive, prohibitively so for many people! The idea behind this amino acid is that it contributes to the formation of anti-oxidants, which allow red blood cells to maintain more flexibility.
Other than these medications, management of sickle cell disease comes down to symptom and sequelae control (e.g., pain management during crises), blood transfusions, and bone marrow transplant.
Because blood transfusions play a part in our upcoming journal club, let’s have a moment for them. However, in case the name didn’t tip you off, this is a pharmacy blog. So we’re not gonna get too crazy deep into this non-medication therapy, but it’s important to have some baseline knowledge.
Blood transfusions have been used in sickle cell disease for ages because, duh, it seems to make sense to give anemic patients some blood. But of course it has to be more complicated than that.
While the aim is to reduce the percentage of red blood cells with HbS and increase the percentage with normal HbA, which will hopefully improve oxygenation and viscosity, blood transfusions come with multiple risks. These include hyperviscosity (if you overshoot and increase hematocrit too much), alloimmunization (aka the patient’s immune system starts a war with the infused blood), iron overload (managed with chelators), and delayed hemolysis.
To try and mitigate some of these issues, patients may receive exchange transfusions rather than simple transfusions. While simple transfusion is (as expected) the infusion of red blood cells on top of what the patient already has, an exchange transfusion removes the patient’s own blood while infusing the new, normal hemoglobin blood.

In summary, the type of transfusion used can affect outcome.
Alright, now that we have the basics out of the way, let’s get ready to RUUUUMMMMBLEEEEE.
Erm. Or just journal club.
A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease: The HOPE Trial
Purpose: To characterize efficacy and safety of voxelotor compared to placebo in patients with sickle cell disease. Pretty standard for a phase 3 trial (phase 1 and 2 trials were already done to investigate in vitro effects, pharmacokinetics, and toxicity).
So what is this voxelotor?
Remember we discussed above how the polymerization of deoxygenated HbS is what leads to sickling of the erythrocytes and all the subsequent clinical complications?
Voxelotor was developed to inhibit that HbS polymerization and stabilize the oxygenated HbS form. In those early trials, it seemed to do a good job of decreasing sickling, decreasing blood viscosity, and extending the life of red blood cells, preventing quite so much hemolysis and anemia.
Theoretically, by increasing the stability of HbS bound to oxygen, it should also mitigate vaso-occlusion and improve transit of red blood cells through blood vessels by keeping those cells out of a sickle state. That being said, one could also imagine that extending the life of red blood cells and improving anemia could lead to increased viscosity (like a simple transfusion can) and risk of vaso-occlusive crisis.
It’s also possible that by stabilizing the binding of oxygen to hemoglobin, the tissues won’t receive the oxygen they need…leading to increased erythropoietin and reticulocyte levels as well as possible ischemia and downstream complications. Log away these thoughts for later.
So, lots of considerations on both the risk and benefit sides. But how do these actually play out in clinical practice?
That’s the whole purpose of this trial.
Funding: The study was funded by Global Blood Therapeutics, who is the maker of voxelotor. According to their website, they launched in 2012 and are currently working on two different sickle cell products, so they are quite specialized. Pfizer actually acquired the company in 2022 and is continuing to develop these products. On the one hand, you could shake your head and say, “How am I supposed to believe anything that’s in this study paper when the study was funded by the manufacturer?”
But let’s be real for a moment… who else is going to fund this study?

It’s ok. Be suspicious. Not dismissive, just wary. (Image)
It’s quite common for this to be the scenario for new drugs (and especially new classes of drugs like this one), and what that means is that we have to assess the information with a fine-tooth comb and a wary eye.
But that doesn’t mean we shouldn’t take anything away at all!
It’s important to note the degree of involvement the sponsor had in the study, which in this case was provision of the study agent and collaboration on study design, data analysis, and data interpretation.
Not to mention there was a sponsor-funded writing assistant that helped the authors prepare the manuscript.
Soooo pretty heavily involved…in fact, that’s about as “involved” as a study sponsor can possibly get. They’re analyzing and interpreting the data, AND they’re writing the manuscript.
BUT, the authors also note in the report that the study was in accordance with all established guidelines AND a contract research organization (IQVIA) and independent data and safety monitoring board reviewed the information along the way.
It’s also rare to see sponsor involvement laid out so transparently. It kind of gives a “we don’t have anything to hide” impression, doesn’t it?
So although the manufacturer was heavily involved throughout (I mean, it is one of two products they’re working on, so that’s only to be expected), they did seem to take extra precautions to ensure the validity of their findings and reporting. So don’t just throw out the study because of its funding source!
Design: This was an international, multicenter, randomized, placebo-controlled, double-blind, parallel-group trial. Well that’s a mouthful. Let’s break this down.
So international and multicenter. Yay, right? That’s what we want to hear on all our randomized controlled trials (RCTs). But rather than breezing over this, take a moment to appreciate that this helps us to know that an attempt was made to make this study all inclusive, regardless of ethnicity or economical background. Now, where was the study actually conducted?
There were 73 study locations, most of which were the 50 United States. Nevertheless, the remaining third of sites included Egypt, France, Italy, Turkey, Kenya, Oman, Lebanon, and the Netherlands, to name a few. So it was actually relatively well-dispersed! We shall investigate later to see if this design was executed in patient numbers or not.
Randomized, placebo-controlled, double-blind, and parallel-group…blah blah blah. Except so NOT blah blah blah! This is HUGE!
These measures are describing to you the efforts that were made to reduce bias throughout the study. It was randomized to reduce selection bias and achieve “equal” (or at least balanced) groups at baseline in terms of demographics and clinical characteristics.
That helps to tease out the effect of the treatment. If we have two groups made up of the same demographics at the start of a study, and we give one group a study drug and one group placebo, then any effect seen in the treatment group is more likely to be due to the drug. We’ll investigate later to see if this played out or not.
It was placebo-controlled and double-blinded to try to minimize influence or bias from patients or providers when evaluating effects of the treatments. And it was parallel-group (as opposed to, for example, a cross over) design, such that patients randomized to a specific group stayed in that group, and the groups were evaluated concurrently rather than in sequence.
You can read more about study design and literature interpretation in Part IV of our Biostats Series.
Inclusion Criteria: Adolescent and adult patients aged 12 - 65 years with confirmed sickle cell disease (multiple variants allowed) were included. They were anemic with baseline hemoglobin levels of 5.5 - 10.5 g/dL and had a history of at least one and up to 10 vaso-occlusive crises in the past year. Pretty characteristic of our sickle cell disease population in the U.S., so that’s off to a good start with regards to external validity.
It will, however, be important for us to see if they characterize the patients by any other comorbitidies or chronic manifestations of sickle cell disease that could influence their disease experiences.
They also included patients who were receiving stable doses of hydroxyurea prior to enrollment. This is important since that means it may have a role as an adjunct therapy rather than having to be used only in treatment naive or refractory patients. We’ll have to add this to our list of things to look at in the results.
Given hydroxyurea is a treatment in and of itself that has proven beneficial in reducing symptomatology, we’ll also need to look at this later to see if groups were balanced in this respect. If one treatment group had a higher number of patients on hydroxyurea at baseline and then a higher level of hemoglobin oxygenation or lower number of sickle cell complications comparatively, how are we to know whether that’s the hydroxyurea or the voxelotor? So just keep that in mind.
Exclusion Criteria: Patients who regularly received exchange transfusions or had gotten one in the last 60 days were excluded. This is a little sad for external validity because it might mean that the more severely impacted sickle cell patients may not have been assessed as candidates, so it may be difficult for us to know whether that subset of people could derive benefit. But that also might make it easier for the researchers to find benefit.
It’s also arguable that the patients who regularly receive exchange transfusions are extra “protected” against crises, at least for some amount of time while their HbA percentages are up, so it might actually be harder to find benefit of a new therapy if they’re not as predisposed to an event. So in that regard, it makes sense that these people were excluded.
It also had to have been at least 2 weeks from the last vaso-occlusive crisis, which makes sense in order to avoid overlap with previous therapies, have patients be in a stable window, and/or be able to tease out the effect of voxelotor alone.

How official study randomization is done... (Image)
Methods: Patients were randomized in a 1:1:1 fashion to voxelotor 1500 mg PO daily, voxelotor 900 mg PO daily, or placebo.
The screening period was 28 - 35 days, treatment lasted for up to 72 weeks (pretty long! Almost 1.5 years of follow up planned!), and then follow up occurred at 3 - 5 weeks after the last dose of intervention (drug or placebo).
Patients were stratified according to hydroxyurea use, age, and geographic location.
Given each of these characteristics could influence either the effect of voxelotor or level of/access to care or sickle cell disease genotype, it makes sense that these were the stratification factors. They will likely help us to be able to tease out what the results would be like in our own respective populations.
Primary Endpoint: The primary endpoint of this study was the percentage of participants who had a hemoglobin increase from baseline of more than 1 g/dL at week 24. Sigh. Why are we a little sad here?
Even though seeing an increase in hemoglobin could be exciting since that could mean decreased hemolysis and sickling in the presence of voxelotor, this is kind of a surrogate endpoint. What really matters to these patients is decreasing acute and chronic complications of sickle cell, such as pain crises or acute chest syndrome, etc.
What we have is a marker that, when it goes up, may indicate improved protection against those complications given the MOA of the drug. BUT they didn’t actually study the complications as their primary endpoint.
So why can’t I be mad in addition to being sad?
Following up on a clinical outcome rather than a laboratory outcome requires more clinical input and expertise, more assessment, likely more time, and therefore more money. So as much as we wish it had been a hard clinical outcome measure (and almost 1.5 years should have provided a decent amount of data in a patient population that highly utilizes the health care system due to crises), we guess we’re not all that surprised that it’s a surrogate. Doesn’t mean we’re not a smidge disappointed though.
Secondary Endpoints: These included the change in hemoglobin from baseline to week 24, markers of hemolysis (including indirect bilirubin, absolute and percent reticulocytes - aka immature red blood cells, and lactate dehydrogenase), erythropoietin levels, and (DRUM ROLL) the rate of vaso-occlusive crisis.
So THERE it is!!
Wish it was the primary endpoint, but that may not have been feasible for the company to pull off time/resources-wise with adequate power. We’d bet the sample size would’ve had to have been much much larger to see a significant different in a clinical endpoint like that than the surrogate endpoint of hemoglobin.
Stats: The primary analysis was done based on intention to treat (ITT), meaning every person who was randomized was included in the analysis according to their randomization group.
This increases the external validity of the trial since it’s more of a real-life picture of the effect. If patients deviated from the protocol, didn’t have 100% adherence with their PO medication, or dropped out, they were still included and analyzed.
There were two pre-specified interim analyses, for which they used the Lan-DeMets alpha-spending function to maintain a type I error rate of 5%.
Huh?
What you should know about this is that when interim analyses are conducted during a study, type I error can be inflated. This means that the chance of falsely rejecting the null hypothesis (and finding a significant difference when there may actually be none) is increased with multiple analyses. So the Lan-DeMets function attempts to account and control for this potential inflation to avoid false conclusions of intervention effects.
As far as analysis of the primary endpoint, the authors used the Cochran-Mantel-Haenszel test. This seems appropriate given the categorical data of hemoglobin response or not (aka, a binary outcome, yes or no) in conjunction with the stratification factors at randomization of hydroxyurea use, geographic region, and age.
Finally, it’s important to note that the authors went with a “worst-case scenario” approach if patients received other therapies after randomization that could have impacted their hemoglobin measures. So if the patients received red-cell transfusions or started hydroxyurea after baseline randomization, they inputted the last known hemoglobin before the transfusion or counted the patient as a non-responder for the primary analysis.
Basically, they didn’t want to take any chances of someone saying it wasn’t their voxelotor that was responsible for a hemoglobin response, so they called any other possibilities “non-responders” to be most conservative.
Baseline Characteristics: In a little under 1.5 years, they enrolled 274 participants at 60 institutions in 12 countries. The three treatment groups were generally well-balanced (and although they didn’t report p-values for comparing between groups, they did state there weren’t significant differences in baseline characteristics). So who were the patients?
Most patients were young Black men and women in their 20s, generally balanced half and half across male and female. Patients actually did span a wide range of geographic regions with ~40% being from non-American, non-European locations. Most patients had homozygous HbS sickle cell anemia with a baseline hemoglobin level of ~8 - 9 g/dL. Over half of the patients had had more than one vaso-occlusive crisis in the past year, although they didn’t break out that range any further than to categorize it as 2 - 10. Over half of the patients were receiving hydroxyurea at baseline.
Overall, pretty characteristic of our typical sickle cell anemia patients in the United States. To answer some of our previous follow up questions, it was well-balanced across groups without any obvious drivers of potential response to treatment. The baseline characteristics seem to engender a good sense of external validity in terms of being representative of the patients we frequently encounter.
On the other hand, however, it WOULD have been nice if they could have further characterized these patients according to co-morbidities and complications of their sickle cell disease. And it WOULD have been helpful to have broken out how many patients had, say, two vaso-occlusive crises in the last year versus 10. Just sayin’, that’s a BIG difference in health care utilization and severity of disease!
It’s also important to note that they reported their baseline characteristics in terms of median and range rather than mean and standard deviation. This should be an indication to us that either the groups were too small to achieve a normal distribution or they had some outliers that would have skewed any reporting of means.
It’s hard to know since they gave us ranges instead of interquartile ranges, the latter of which at least lends some sense of distribution of the range.
For example, when they say that the baseline hemoglobin ranged from 5.9 to 10.8 in the voxelotor 1500 mg group, we have no way of knowing where within that range the majority of people fell. Did just one person have very low hemoglobins, or was it several? We can’t really tell here, which leaves us a bit in the dark as to interpretation of potential response in varying severity subsets of patients.
Follow up was of a decent duration, largely ~40 weeks for all groups according to their medians. 80% of a year should have been decently sufficient to see patterns in vaso-occlusive crises.
But oh wait, they were looking at hemoglobin…not vaso-occlusive crises. They also gave those durations of follow up as ranges with some patients being followed for much shorter periods of time after enrollment (Only 1-2 patients in each group actually made it all the way until the 72 week mark, so that planned 1.5 year follow up didn’t really play out).
Efficacy Outcomes: The pre-defined hemoglobin response at week 24 in the ITT analysis was seen in 51% of the voxelotor 1500 mg group, 33% of the voxelotor 900 mg group, and only 7% of the placebo group. There was a significant difference between 1500 mg and placebo with p < 0.001. So even though there is no reported power analysis in this study, because a significant difference was found, a significant difference was found. Power’s out the window on the primary endpoint, we already have significance.
These results were consistent in the per-protocol analysis, as well as in patients who were taking hydroxyurea or had severe anemia at baseline. Using the ITT group, the number needed to treat (NNT) to produce that hemoglobin response comes out to be 2.3, so with conservative rounding, about 3 patients. That’s a super low NNT!
Interestingly, changes in hemoglobin and hemolysis markers were evident in the first 2 weeks of treatment, which seems to indicate a rapid response that was largely maintained throughout therapy with voxelotor (see below).

(Image)
Markers of hemolysis, including decreased indirect bilirubin, absolute reticulocyte count, and lactate dehydrogenase, were also improved in the voxelotor 1500 mg group compared with the placebo group, although not all of these were statistically significant.
The fact that neither reticulocytes (nor erythropoietin) were increased in the voxelotor group also seems to indicate that tissues weren’t oxygen-hungry.
It’s also important to note that the percentages of patients who had red-cell transfusions during the trial were similar between all three treatment groups, indicating that this treatment modality was balanced across groups and can not be implicated in interpreting differences in treatment safety or efficacy.
Now to the clinical meat of the study, what we all came for (even though it’s not the primary outcome…)! The rates of vaso-occlusive crises. The annualized incidence rates of vaso-occlusive crises (aka crises per person-year) were 2.77 in the voxelotor 1500 mg group, 2.76 in the voxelotor 900 mg group, and 3.19 in the placebo group.
These rates were adjusted for hydroxyurea use, geographic region, and age, such that the differences in crises rates could not be interpreted as differences in those stratification factors. Sooo, what do we think of this? 2.77 crises per person-year versus 3.19? Not really a huuuuge difference from a clinical perspective.
On the one hand, voxelotor didn’t really seem to make a difference in the numbers of these crises for patients (query: how much of a difference will it make from a clinical perspective?). But on the other hand, it didn’t really seem to make a difference in these numbers (so it at least doesn’t seem to be making rates of vaso-occlusion worse by making HbS hold on to the oxygen more).
Two sides of the mirror, different lenses to look through for efficacy and safety.
Also, are those numbers not really that different because there’s really not a difference, or because this study wasn’t powered for or run long enough to find a difference?
Now let’s look at the slightly more severe subgroup of patients who already had at least two crises in the past year prior to randomization. Those patients’ incidence rates were 2.88 in the voxelotor 1500 mg group, 3.39 in the voxelotor 900 mg group, and 3.5 in the placebo group. Now that’s looking a little more clinically meaningful to save almost a whole crisis per person-year!
When you figure each vaso-occlusive crisis admission is often at least a week’s duration, to save a person at least one whole week in the hospital per year (and all the possible complications and costs that go along with that) is a bit more meaningful.
Safety Outcomes: Adverse event rates were similar across the three treatment groups with the most commonly reported events being grade 1 or 2 headache and diarrhea. Pretty minor on the spectrum of adverse events. There was no difference in more serious grade 3 adverse events between groups. Four participants did pass away during the study, but these were deemed to be unrelated to the trial or drug by the investigators.
Authors’ Conclusions: Voxelotor did what it was supposed to do. It increased hemoglobin levels, improved anemia, and reduced hemolytic markers. More info is needed for determining how effective it is for preventing vaso-occlusive crises, which is ongoing in a open-label extension study. Yay!
tl;dr Conclusions: Tbh, we can’t really argue with the authors’ conclusions. The medication set out to do what they wanted it to do in a wide population range with variable disease severity and other treatments, and it did so with relatively few adverse effects. It’s just as yet unclear whether it will have a huge clinical impact on patients’ lives.
We really need a lot more information about clinical effects rather than just laboratory effects before we can make any solid conclusions about incorporating this into practice. Hellooooo extension study! Which, as we now know in 2024, was a big ol’ bust. Womp womp.
Not to mention, we need info about the financial impact of voxelotor. Hydroxyurea is ~$1.30/500 mg capsule (cash price), which is really quite affordable. Endari, on the other hand, is ~$22/5 g packet. Where will voxelotor fall on the price spectrum? I can’t imagine it’ll be cheap, but remember that length of stay for a vaso-occlusive crisis and all of THOSE costs...
Buh BAM. Journal club time is over - for now. We HOPE (see what we did there?) this has given you some insight into reading an article and trying to come away without the wool pulled over your eyes. Because when the providers come talk to you about prescribing this cool drug that sounds like the newest Transformer, you’ll want to be informed. Read and discuss amongst yourselves. You’ve read our thoughts, what do YOU think?

Oh, you thought this was Optimus Prime? WRONG. It’s just voxelotor about to whoop some hemoglobin S @$$. (Image)