
The Pharmacist's Guide to Sickle Cell Disease
Editor's note: Cara Clayton is a P4 student from the University of Texas at Austin College of Pharmacy. She is interested in Oncology and Pediatric Pharmacy and hopes to attain a PGY1 residency to further her clinical knowledge and become more involved in the patient care process. Pending successful completion of a PGY1 residency, she hopes to pursue a PGY2 residency in Oncology and become a Board Certified Oncology Pharmacist.
She was kind enough to take an area that pharmacists don't really learn about (sickle cell disease), and give it a tl;dr judo-chop. I learned a ton reading this article, and I'm sure you will too. Thanks Cara!
In most Pharmacy curriculums, sickle cell disease is mentioned maybe once or twice (or not even at all). This makes sense when you consider the number of more prevalent disease states (diabetes, hypertension, dyslipidemia, etc...) that we have to learn about in a mere 4 years.
For most pharmacists, encounters with sickle cell disease are few and far between. However, if you work in a city (particularly if it's an international hub such as DC or New York), you will encounter sickle cell somewhat regularly.
On top of that, if you have any interest in heme/onc or pediatrics, sickle cell disease is something you will need to know.
So, with all that said, let's jump in and learn about the (not so) wonderful world of sickle cell disease.
Sickle Cell Disease Background
The term sickle cell disease (SCD) encompasses a group of inherited disorders that affect hemoglobin (the molecule in our red blood cells (RBCs) that selflessly delivers oxygen to all of our tissues).
Individuals with SCD have abnormal hemoglobin molecules, called hemoglobin S. These atypical hemoglobin S molecules distort RBCs into a crescent, or sickle, shape.

Visual of hemoglobin (Image)
As a refresher from first year biochemistry (I can already hear the groans), hemoglobin is found in our RBCs and is made up of 4 heme molecules, plus 4 protein subunits called globulins (hence the name hemoglobin).
Each heme molecule also has 1 iron molecule that can bind to 1 oxygen molecule.
For the protein part of hemoglobin, 2 of the subunits are called alpha globulin, and 2 are called beta globulin.
As you might be able to guess, mutations in the gene that codes for alpha globulin (hemoglobin subunit alpha [HBA] gene) result in various version of alpha globulin. And vice versa for beta globulin (only this time, the gene that codes for it is...wait for it...hemoglobin subunit beta [HBB] gene).
Zooming back out, the globulin proteins (2 alpha and 2 beta) hold the various heme molecules in place. Oxygen binds to that iron molecule in heme, and takes a ride on an RBC through your circulatory system until the oxygen eventually is deposited at a tissue in need.

Visual of the probability of inheritance of SCT and SCD if both parents are carriers of SCT (Image)
SCD is an autosomal recessive trait. This means it is inherited (it's not contagious, and you cannot catch it).
People who inherit one sickle cell gene and one normal HBA gene have the sickle cell trait (SCT).
Individuals with SCT typically do not have symptoms of SCD, but they can pass the trait on to their children (and they may have some protection against malaria as a nice side-benefit).
If both parents carry SCT, their children have a 1 in 2 (50%) chance of also having SCT, and a 1 in 4 (25%) chance of being born with SCD.
SCD is particularly common among people whose ancestors came from sub-Saharan Africa, Spanish-speaking regions in the Western Hemisphere, Saudi Arabia, India, Turkey, Greece, and Italy.
The CDC estimates that:
Approximately 100,000 Americans are affected by SCD
SCD occurs in about 1 out of every 16,000 Hispanic-American births
About 1 in 13 African-American babies is born with SCT
People who have SCD inherit one abnormal HBB gene from each parent. In every form of SCD, at least one of the two beta globulin mutations results in the sickle mutation, which then produces hemoglobin S.
Homozygous SCD is a result of inheriting two copies of hemoglobin S, and is referred to as sickle cell anemia. Heterozygous SCD is a result of the sickle mutation plus another beta globulin mutation. These other beta globulin mutations result in other abnormal forms of hemoglobin, such as hemoglobin C, hemoglobin D, and hemoglobin E.
Some forms of SCD include:
Hemoglobin SS (HbSS)
Hemoglobin SC (HbSC)
Hemoglobin Sβ0 thalassemia (HbS-beta0 thalassemia)
Hemoglobin Sβ+ thalassemia (HbS-beta+ thalassemia)
Hemoglobin SD (HbSD)
Hemoglobin SE (HbSE)
Sickle cell anemia is the most common form of SCD, and frequently the most severe. Other common forms of SCD include hemoglobin SC disease and hemoglobin Sβ thalassemia.
In terms of diagnostics, SCD diagnostic testing includes prenatal testing and newborn screening. Since pharmacists are not commonly involved with diagnostics, I will not spend much (read: any) time covering this.
If you want more info on the diagnostic testing of SCD, you can read up on it here.
Sickle Cell Disease Pathophysiology
As we know, cells in our tissues need a constant supply of oxygen in order to work like they should. RBCs with normal hemoglobin are disc shaped and flexible enough to move through large and small blood vessels to deliver oxygen.
But, hemoglobin S is not like normal hemoglobin. As a result, it forms stiff rods in the red blood cell, changing it into a sickle shape.

A depiction of the different shapes of sickle cells and normal red blood cells
These sickle-shaped cells lack the flexibility of normal RBCs and can stick to vessel walls, resulting in a blockage that slows or stops blood flow.
When blockages occur, oxygen cannot reach nearby tissues. You probably don't need me to tell you this, but your tissues get upset when they don't have oxygen.
The end result is vaso-occulsive pain crises, or attacks of sudden, severe pain.
These pain crises can occur without warning and often require hospitalization for effective treatment. Over time, poor oxygen delivery resulting from SCD can also harm organs such as the kidneys, lungs, eyes, liver, heart, and brain.

An image of sickled cells resulting in blockages
Additionally, sickle cells tend to hemolyze (i.e. burst apart into thousands of pieces), since they cannot change shape easily.
This makes it hard for the body to keep up with RBC production demands...old RBCs are destroyed faster than new ones can be made. This high turnover rate results in anemia.
Some of the downstream complications of SCD include
Vaso-occlusive pain crises
Acute Chest Syndrome
Bone and joint complications
Cerebrovascular disease
Renal, Hepatic, and Pulmonary disease
Priapism
Pulmonary hypertension
Sickle Cell Disease Treatment
SCD is a life-long illness. In high-income and developed countries, like the US, the life expectancy of a person with SCD is about 40-60 years.
Currently, the only cure is a hematopoietic stem cell transplant (HSCT).
There are 2 kinds of HSCT:
Allogeneic - Uses stem cells from an HLA-matched donor
Autologous - Uses a person's own stem cells
That HLA above stands for "Human Leukocyte Antigen." It's a protein that lives on your macrophages, and it's involved in telling your immune system what is "self" and what is "not-self."
As you probably remember, its the job of your immune system to remove "not-self" things from your body. So it's really important with an allogenic transplant that the HLA types match up. Otherwise, the patient will reject the transplant.
With autologous HSCT you don't have to worry about HLA so much since you're transplanting a patient's own cells back into them.
Anyway, for SCD patients, allogeneic transplants are far more common. Also, only young patients (< 16 years old) with SCD are eligible for HSCT due to an increased risk of severe toxicities and death among individuals > 16 years old.
Due to the lack of a widely available cure, SCD management is primarily focused on preventing and treating complications.
There are a few main areas that we focus on for SCD patients:
Infection Prevention
Nutrition
We'll cover them all in more detail below.
Infection Prevention in Sickle Cell Disease
SCD patients are highly susceptible to bacterial and viral infections. Our strategies to prevent these are immunizations and prophylactic antibiotics. Specifically with SCD patients, we'd consider using prophylactic penicillin therapy in patients < 5 years old.
Children with SCD should receive all routinely recommended childhood vaccines. Additionally, they should receive both the pneumococcal conjugate (PCV13) and pneumococcal polysaccharide (PPSV23).
PCV13 can be administered as young as six weeks of age and should be given in four doses. PPSV23 must be administered after 24 months of age because it is not immunogenic in children younger than two years of age.
The recommended administration schedule of these vaccines can be found below.
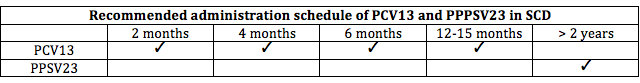
Sickle Cell Disease Vaccination Schedule
In addition to vaccines, penicillin prophylaxis should be used in all patients with SCD until the age of five to minimize risk of Streptococcus pneumoniae infection.
The prophylactic dose for patients 3 months to 3 years of age is 125 mg penicillin VK by mouth twice daily, while the prophylactic dose for patients 3 years to 5 years of age is 250 mg penicillin VK by mouth twice daily.
3 months - 3 years: 125 mg penicillin VK BID
3 years - 5 years: 250 mg penicillin VK BID
In the case of a penicillin allergy, the patient should receive prophylactic erythromycin twice daily.
Nutrition in Sickle Cell Disease
Growing evidence suggests patients with SCD have vitamin and micronutrient deficiencies that can influence the course of their disease. Thus, the following supplements are recommended:
Folic acid: This should be given to all patients with SCD as 1 mg by mouth once daily, unless the clinician determines the patient has sufficient dietary intake and decides to omit this therapy.
Daily multivitamin without iron: Iron should be excluded because excessive iron stores and oxidative injury can add to the depletion of antioxidant vitamins.
Oral vitamin D and calcium: Recommended only for patients with SCD also found to be vitamin D deficient.
Pain Management in Sickle Cell Disease
Acute vaso-occlusive pain crises are one of the most common reasons for patients with SCD to seek medical attention. Additionally, chronic pain affects a large proportion of these patients.
We can manage pain both reactively (with opioids and other analgesics) and proactively (with preventative measures). We'll discuss both below.
We'll start by talking about hydroxyurea, whose role in SCD extends far beyond pain management (but it's important there too).

That's an awful lot of benefits from one little pill...(Image)
Hydroxyurea is an important mainstay in the overall management of patients with SCD due to its ability to prolong survival, decrease the incidence of acute painful episodes, and reduce hospitalization rates.
So, how does it work?
Spoiler alert: you are about to find out!
Hydroxyurea is an antimetabolite that selectively inhibits ribonucleoside diphosphate reductase.
This means it is essentially oral chemotherapy.
This is very important to remember for real-life pharmacy practice. Hydroxyurea has not one, but two black box warnings due to its carcinogenic properties and ability to cause severe myelosuppression.
Because of this, hydroxyurea must be handled and counseled on extremely carefully.
The National Institute for Occupational Safety and Health (NIOSH) recommends health care workers wear single gloves during receiving, unpacking, and placing in storage, and double gloves with a protective gown when manipulating tablets/capsules to prepare an oral suspension.
Not just any gloves, mind you. But ASTM-rated gloves that are resistant to penetration by chemotherapy (in a healthcare institution, you'll probably just call these "chemo gloves").
Basically, don't touch hydroxyurea bottles, tablets, or even the box it came in with your bare hands. That goes for pharmacists/techs receiving and dispensing the drug, and it goes for nurses administering the drug.
On the outpatient side, it's still a best practice for the patient to wear gloves when handling the tablets (although since they are swallowing the tablets anyway you may be met with some resistance here).
At a minimum, ensure they know to wash their hands after handling hydroxyurea bottles or tablets.
But their family members absolutely need to wear gloves if they are touching the tablets (and the patient should be advised to keep hydroxyurea out of reach of children, pets, etc).
What else do patients need to know?
They need to use sun protection (because hydroxyurea can cause photosensitivity) and they need to keep their follow-up appointments with their hematologist (because hydroxyurea can cause secondary malignancies).
Because of hydroxyurea’s ability to cause myelosuppression, blood counts must be monitored at baseline and throughout treatment. Fortunately, if hydroxyurea-induced myelosuppression occurs, it's usually reversible with treatment interruption.
In instances of severe bone marrow depression, hydroxyurea must be held. In the setting of SCD, "severe bone marrow depression" is defined as:
Neutrophils < 2,000/mm3
Platelets < 80,000/mm3
Hemoglobin < 4.5 g/dL
Reticulocytes < 80,000/mm3 when hemoglobin is < 9 g/dL
So, now that you know the scary information about hydroxyurea, you may be wondering about how it works in SCD.
Since it's an antimetabolite (remember that old ribonucleoside diphosphate reductase inhibition bit from above?), hydroxyurea slows the production of sickle hemoglobin and increases production of fetal hemoglobin.
It can also wield beneficial effects on the hydration of RBCs, vascular wall adherence, and suppression of granulocyte (white blood cells) and reticulocyte ("pre-RBCs") counts.
For SCD, hydroxyurea is dosed in a range of 15-35 mg/kg/day and titrated by 5 mg/kg/day every 12 weeks to maintain a white cell count between 5,000 and 8,000 cells/mm3.
As mentioned above, if toxicity occurs, treatment must be held until bone marrow recovers. After bone marrow recovery, treatment can be restarted with a dose reduction of 2.5 mg/kg/day and titrated up by 2.5 mg/kg/day every 12 weeks as long as no additional toxicity occurs.
If hematologic toxicity occurs a second time, hydroxyurea therapy should be permanently discontinued.
Does every patient with SCD get hydroxyurea?
No. As we've seen above, although there are some great benefits to hydroxyurea, there are also a lot of risks.
Generally speaking, hydroxyurea is considered an option in patients who experience > 3 vaso-occlusive pain crises per year requiring hospitalization, or repeated episodes of acute chest syndrome (more on acute chest syndrome in a minute).
Circling back to pain management, hydroxyurea's benefits for preventing pain typically do not surface until three months after starting therapy.
Thus, if a hydroxyurea-naive patient is hospitalized with an acute pain crisis, you're probably not going to start them on it in the hospital without consulting their primary hematologist. You'll resolve the pain crisis first, and try to coordinate starting hydroxyurea as an outpatient if the hematologist is on board.
Aside from hydroxyurea (used to prevent pain crises), the management of an acute vaso-occlusive pain crisis includes:
Analgesics
Hydration
Evaluation for underlying causes
As discussed above, SCD causes obstruction in the microvasculature, which results in tissue ischemia and inflammation.
That ischemia and inflammation is what leads to the pain crisis, and it's often precipitated by triggers such as dehydration, insomnia, overexertion, asthma exacerbations, and hormonal changes.
Acute pain management in a vaso-occlusive crisis should be initiated early and aggressively (yes, that means opioids).
There isn't a "one size fits all" approach to pain management with SCD patients. The plan and goals should be individualized for each patient.
Patients with SCD frequently get a bad rap for abusing or depending on opioids, but this is often unjustified. The pain from an acute pain crisis is very real and needs to be managed.
Unfortunately, opioids are very prone to tolerance and dependence, so trust between the patient and clinician is extremely important.
For pharmacists, we can help by counseling on the side effects of opioids (don't forget to start a bowel regimen to prevent opioid-induced constipation!) and information on accessibility of opioid reversal agents (i.e. naloxone).
During hospitalization for acute pain crises, continuous opioids via patient-controlled analgesia (PCA) may be needed.
Hydration is important because SCD patients are often hypovolemic during acute painful episodes. This is due to decreased oral intake, increased insensible losses, and reduced urinary concentrating ability of the kidneys.
Hypovolemia is initially treated with a bolus of 0.5 – 1 L of normal saline (NS), followed by a maintenance rate of combined oral and IV fluids targeted to a net even or slightly positive daily fluid balance.
Because patients with SCD appear to be hypercoagulable at baseline, VTE prophylaxis should be initiated in all adults (i.e. patients > 18 years of age) with SCD hospitalized for an acute medical illness.
Thromboprophylaxis is recommended with one of the following agents:
A low molecular weight heparin at prophylactic doses (e.g. enoxaparin 40 mg subcutaneously every 24 hours or dalteparin 5,000 units subcutaneously every 24 hours)
Unfractionated heparin 5,000 units subcutaneously three times daily
Fondaparinux 2.5 mg subcutaneously once daily
A while back, I promised we'd talk about Acute Chest Syndrome. Thank you for waiting so patiently! The time has finally arrived.
Acute Chest Syndrome
Acute chest syndrome (ACS) is a leading cause of death for patients with SCD.
The Cooperative Study of Sickle Cell Disease (CSSCD), the largest natural history study of SCD, estimates that 50% of patients with SCD will have an episode of ACS.
The upshot of that is that adults with ACS are more likely to require mechanical ventilation, have a prolonged hospital stay, and have an increased risk of death.
Risk factors for ACS (among adults > 20 years old with SCD) include:
Increased white blood cell count
Increased hemoglobin S levels
Decreased fetal hemoglobin levels
Smoking
Vaso-occlusive pain crises
And some of the causes of ACS include bone marrow and/or fat emboli, infection, asthma, compromised ventilation, and pulmonary thrombi, as seen by this lovely picture below.

(Image) Pathophysiology of ACS and vaso-occlusive pain crises
ACS commonly presents as chest pain and shortness of breath, and about 50% of ACS patients have pain episodes preceding the ACS event.
ACS is diagnosed by the presence of a new segmental radiographic pulmonary infiltrate (this link will tell you WAY more than you need to know about interpreting chest X-Rays, but it's super informative) and at least one of the following:
Temperature ≥38.5°C
>2% decrease in O2 saturation from a documented steady-state value on room air
PaO2 < 60 mmHg
Tachypnea
Intercostal retractions, nasal flaring, or use of accessory muscles of respiration
Chest pain
Cough
Wheezing
Rales
ACS severity is defined as mild, moderate, severe, or very severe (diagnostic criteria below):

Acute therapy for ACS includes:
Fluid management to prevent hypovolemia
Supplementary oxygen
Blood transfusion
On the preemptive side, we can use hydroxurea, chronic transfusion therapy, and HSCT can help to prevent ACS.
Finally, while not FDA approved for pain associated with SCD, adjunctive antidepressants or anticonvulsants may help.
In October 2010, the American Journal of Hematology published a cohort study evaluating the use of adjunctive agents for vaso-occlusive pain in 2,194 patients with SCD.
This study found that use of...
Stronger opioids (morphine and oxycodone)
Antidepressants (SSRIs and SNRIs), or
Anticonvulsants (carbamazepine, valproic acid, phenytoin, gabapentin)
...was significantly associated with lower cumulative rates of acute vaso-occlusive pain visits over time.
So it's possible that you'll see them used adjunctively for SCD patients.
New and Investigational Drug Therapies for Sickle Cell Disease
For the first time in about 20 years (literally), we've got a new player to the SCD treatment landscape.
There's also some investigational stuff in the pipeline, but let's start with the recently approved new drug.
Endari™ (L-glutamine) was approved by the FDA on July 7, 2017 for use in patients > 5 years of age with SCD to reduce severe complications associated with this disorder.
Glutamine is a precursor for nicotinamide adenine dinucleotide (NAD), which has a role in regulating and preventing RBC oxidative damage.
Endari is supplied in a 5 gram oral packet that should be mixed with 240 mL of a cold or warm beverage (e.g. water, milk, juice), or with 120-180 mL of food (e.g. applesauce, yogurt) prior to administration.
Dosing is based on the patient’s weight and is as follows:
< 30 kg: 5 grams (1 packet) by mouth twice daily
30 – 65 kg: 10 grams (2 packets) by mouth twice daily
> 65 kg: 15 grams (3 packets) by mouth twice daily
Potential adverse effects associated with Endari use include chest pain, headache, constipation, nausea, abdominal pain, limb or back pain, and cough.
One 5 gram packet costs $22.20 without insurance, meaning this product can get pricey depending on the patient’s age, weight, and insurance coverage.
To put this in perspective, a 30-day supply of this product for a patient without insurance who weighs < 30 kg would cost $1,332 per month, which equates to $15,984 per year.
Compared to the relatively cheap hydroxyurea (far less than $1.00 per pill), that's an expensive proposition. Cost may initially limit Endari's utility for the time being.
Drugs targeting red cell adhesion and/or leukocyte adhesion are attractive treatment options due to the contribution of adhesive interactions to vaso-occlusion.
See below for a table of ongoing and recently completed studies of therapies targeting adhesion from the Journal of the American Society of Hematology.

(Image)
Selectin inhibitors, or drugs that target selectin-mediated adhesion are being especially actively investigated. These medications inhibit adhesion and/or activation of leukocytes normally recruited to inflamed vessels.
Previous animal studies showed that inhibiting P-selectin-mediated and E-selectin-mediated adhesion led to a reduction of vaso-occlusion (which would potentially lead to reduced complications for SCD patients).
Additionally, drugs targeting inflammatory pathways are of interest due to inflammatory responses caused by vaso-occlusion.
Vaso-occlusive inflammatory responses often resemble hypoxia or reperfusion injuries and involve mononuclear lymphocytes, polynuclear lymphocytes, and the production of proinflammatory cytokines.
See below for a table of ongoing and recently completed studies targeting inflammation from the Journal of the American Society of Hematology.
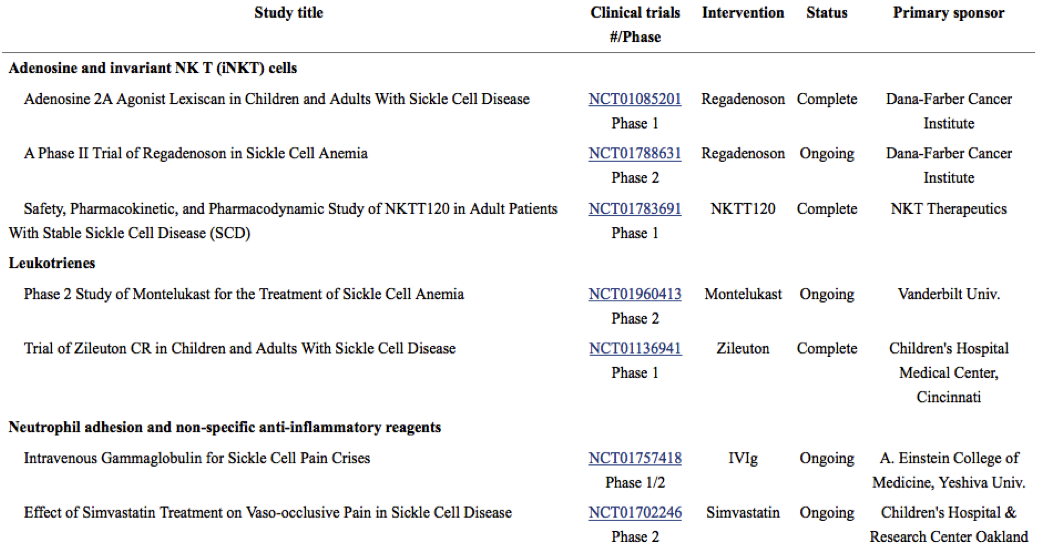
(Image)
This post is already long enough without discussing in detail each new medication in the pipeline, but as you can see, there are many new and old drugs being studied.
While none of these products are ready for FDA approval just yet, the increase in clinical pharmaceutical research directed towards SCD and the number of drugs in the pipeline make it reasonable to expect new treatments for SCD in the near future.